Enfermidade xenética rara que involucra ao cromosoma 18, na cal falta o brazo longo (total ou parcialmente) nun dos pares do cromosoma (deleción terminal), alterando o desenvolvemento do cerebro e algunhas estruturas corporais. Este síndrome adoita ter lugar de forma esporádica e espontánea nas primeiras etapas do desenvolvemento embrionario, afectando aproximadamente a 1 de cada 40.000 bebés que nacen.
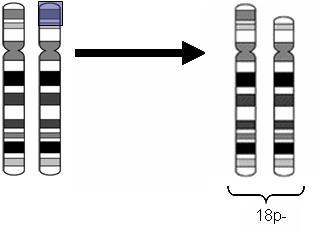
Na forma clásica da enfermidade, o síndrome dismórfico é discreto e non específico. Os signos clínicos principais son:
- estatura baixa,
- cara redonda con filtrum curto,
- ptosis palpebral (A ptosis palpebral é a caída do párpado superior. Xurde, habitualmente, por unha disfunción do músculo elevador, debido a causas dexenerativas ou conxénitas).
- orellas grandes e despregadas.
- O déficit intelectual é de leve a moderado.
- Un subgrupo de pacientes, entre o 10 e 15 %, presentan graves malformaciones cerebro/faciais que evocan ao espectro de anomalías da holoprosencefalia (constitúe un amplo espectro de malformacións do cráneo e a cara debidas a unha anormalidade complexa do desenvolvemento do cerebro que teñen en común a ausencia do desenvolvemento do prosencéfalo, que é o lóbulo frontal do cerebro do embrión. Durante o desenvolvemento normal fórmase o lóbulo frontal e a cara comeza a desenvolverse na quinta e sexta semana do embarazo. A holoprosencefalia é causada pola falta de división do lóbulo frontal do cerebro do embrión para formar os hemisferios cerebrais bilaterales (as metades esquerda e dereita do cerebro), causando defectos no desenvolvemento da cara e na estrutura e o funcionamento do cerebro)
As análises citoxenéticas son necesarias para realizar un diagnóstico definitivo. Os diagnósticos diferenciais poden incluír un gran número de síndromes con:
- estatura baixa e
- déficit intelectual leve.
- En nenos pequenos, a síndrome da deleción 18p pode evocar ligeramente a unha síndrome de Turner ou a unha trisomía 21. A deleción 18p pode detectarse de forma prenatal grazas á análise citoxenético de amniocitos ou células das vellosidades coriónicas. Non existe un tratamento específico, pero a reeducación da linguaxe e o manexo cedo poden contribuír a mellorar a evolución dos nenos afectados. Salvo para os pacientes con malformaciones cerebrais graves, a esperanza de vida non parece estar diminuída de forma significativa.
FONTE: Dra Catherine TURLEAU
Ningún comentario:
Publicar un comentario