 A enfermidade de Pelizaeus-Merzbacher (PMD) é un trastorno xenético raro, ligado ao cromosoma X que afecta o sistema nervioso central e está asociado con anormalidades da sustancia branca do cerebro e a medula espinal.
A enfermidade de Pelizaeus-Merzbacher (PMD) é un trastorno xenético raro, ligado ao cromosoma X que afecta o sistema nervioso central e está asociado con anormalidades da sustancia branca do cerebro e a medula espinal.
É unha das leucodistrofias nas que a enfermidade se debe ao desenvolvemento anormal dun ou máis compoñentes (predominantemente graxas ou proteínas) que forman a sustancia branca (vaina de mielina) do cerebro.
A vaina de mielina é a cuberta protectora do nervio. Os nervios non poden funcionar normalmente sen ela.
 En PMD, moitas áreas do sistema nervioso central poden estar afectadas, incluíndo as porcións profundas do cerebro (subcortical):
En PMD, moitas áreas do sistema nervioso central poden estar afectadas, incluíndo as porcións profundas do cerebro (subcortical):- cerebelo,
- tronco cerebral e
- médula espinal.

Signos e Síntomas
Os signos de PMD poden variar amplamente de persoa a persoa. Os signos da forma clásica de PMD xeralmente comezan durante a primeira infancia, tipicamente antes dos 2 meses de idade. Inicialmente, os bebés afectados poden non desenvolver un control normal da cabeza e os ollos, especificamente movementos anómalos da cabeza e movementos rápidos, involuntarios e sacudidas dos ollos (nistagmo).
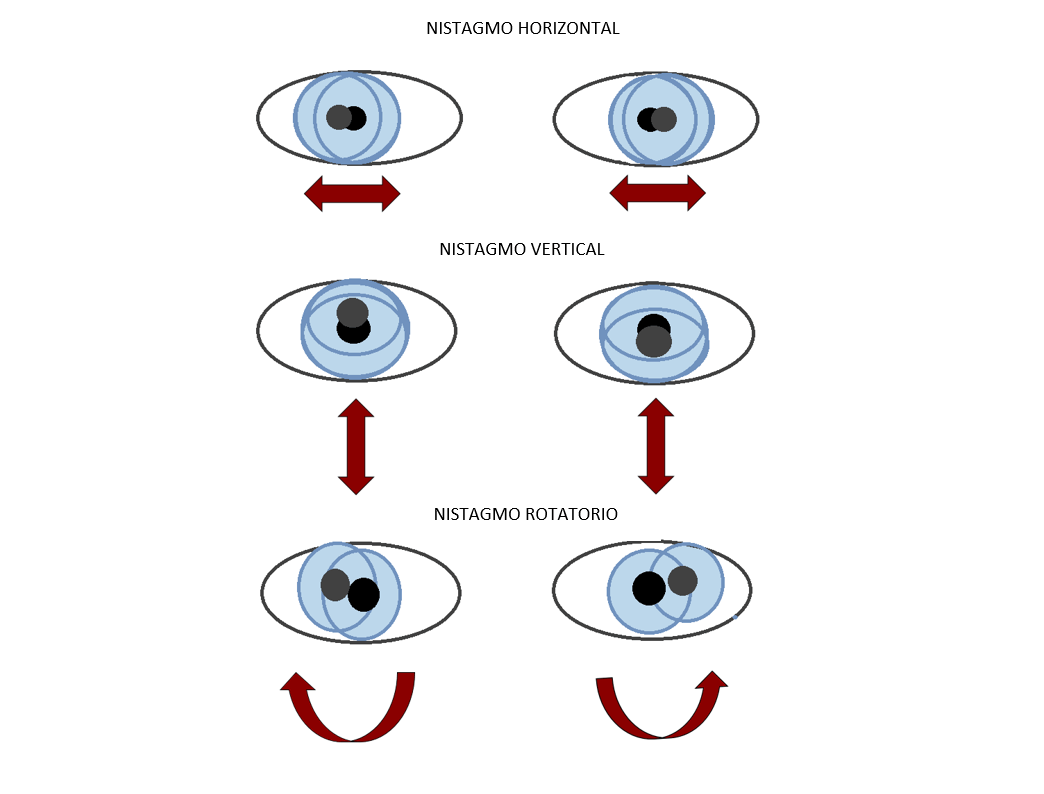

Os signos de PMD connatal están presentes ao nacer ou se observan durante as primeiras semanas de vida. Esta forma do trastorno caracterízase por debilidade, espasticidad, estridor, nistagmo e convulsións. Tamén pode ocorrer unha disfagia grave, que require unha alimentación con gastrostomía. Os nenos afectados tamén poden mostrar deterioración das funcións mentais e o fracaso para alcanzar os fitos do desenvolvemento, como falar e camiñar. A progresión desta forma de PMD é máis rápida e severa que a forma clásica. A miúdo é fatal durante a infancia.
A PMD de transición é unha forma de enfermidade que é intermedia entre as formas clásica e connatal. Os signos son similares aos das formas clásica e connatal do trastorno. Con todo, a velocidade de progresión é máis rápida que a forma clásica, pero máis lenta que a forma connatal.
Causas
PMD hérdase como un trastorno xenético recesivo ligado ao cromosoma X que afecta principalmente aos homes. Os trastornos xenéticos ligados ao cromosoma X son trastornos causados por un xene anormal no cromosoma X. As mulleres que teñen un xene da enfermidade presente nun dos seus dous cromosomas X son portadores dese trastorno. Polo xeral, as portadoras non presentan síntomas porque un dos seus dous cromosomas X está inactivado de modo que os xenes dese cromosoma non funcionan. Normalmente é o cromosoma X co xene anormal que está inactivado. Os homes teñen un cromosoma X que se herda da súa nai e, se un home herda un cromosoma X que contén un xene da enfermidade, vai desenvolver a enfermidade.

Diagnóstico
Pódese sospeitar un diagnóstico de PMD baseado nunha avaliación clínica exhaustiva, unha historia detallada do paciente e unha variedade de probas especializadas como a resonancia magnética (MRI) para detectar a deficiencia de sustancia branca. O recoñecemento de defectos de mielinización temperá, como a falta de mielinización no cerebelo e o tronco encefálico, pode axudar no diagnóstico precoz das formas graves de PMD. Disponse de probas xenéticas moleculares para o xene PLP1 para confirmar o diagnóstico.
Terapias estándar
Tratamento
Non existe un método ou réxime de tratamento estándar para os individuos con PMD. O tratamento baséase en síntomas específicos presentes, como os medicamentos que preveñen as convulsións ou os utilizados para os trastornos do movemento. Recoméndase a atención de apoio, incluíndo apoio emocional para os membros da familia, segundo sexa necesario.
O asesoramento xenético recoméndase para as persoas afectadas con PMD e as súas familias.
{kind=link}
FONTE:
Ningún comentario:
Publicar un comentario